Models of protein sequence
Alex Lee, 2023-10-17
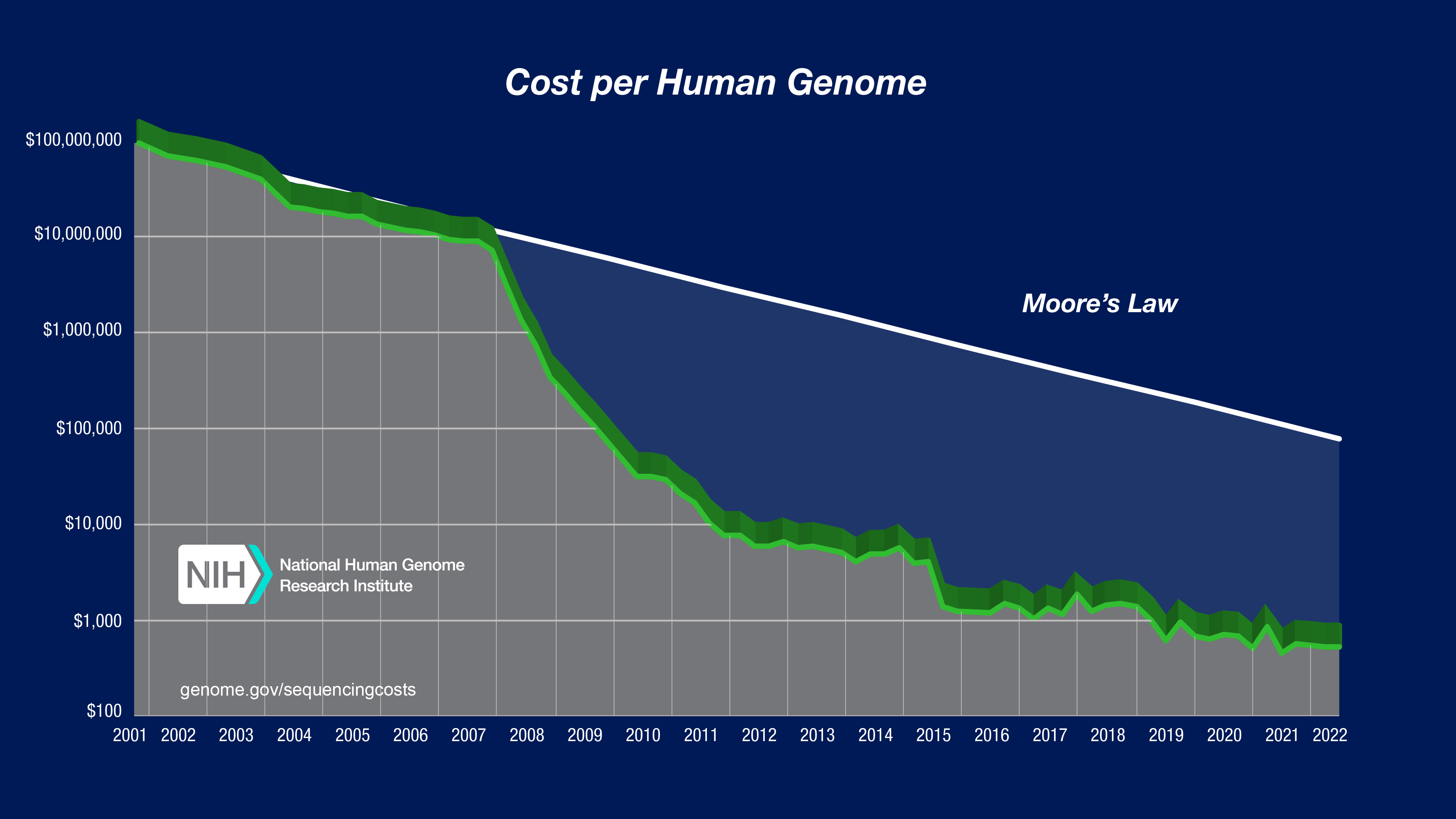
The amount of sequencing data is growing (extremely) quickly
NVIDIA estimates 40 exabytes of data by 2025!–driven by decreases in cost.
Protein sequence models have typically focused on simple probabilistic models of amino acid frequency
The mainstay tool of these analyses has been a multiple-sequence alignment.
- collect sequences corresponding to a protein (across species, individuals, etc.)
- align them using computational approaches (ex DP w/ Needleman-Wunsch etc.)

Conservative/non-conservative terminology refers to (broad sense) notions of similarity across species or between individuals [ie T/S conservative site largely the same across species]
What do mutation sites indicate?

Each ball is an amino acid (AA); AAs colored in orange/purple are the ones in the sequence alignment denoted by different colors; hashes are sites that have high entropy, and are likely to be functionally relevant.
Early models (like in this paper, from 2007) focused on creating probabilistic models of specific sites based on entropy (\(\sum_i p_i\ \mathrm{ln}(p_i)\)) at a given site (column of alignment matrix) and clustering sequences based on their entropy at different sites.
A key assumption of this idea is basically the idea that if a residue (individual AA) is commonly observed in the population then it has high “fitness”. Any statements made are population level about a given sequence alignment.

More sophisticated models and applications to disease
Marks lab at Harvard pioneers more complex probabilistic models based on stat. mech.

Previous method focuses on marginal probabilities at specific sites \(\mathrm{\mathbf{h}_i}(\sigma_i)\) [ie a fixed effect] for a given protein alignment.
Now we have an overall model of a given sequence \(\sigma\): \(P(\sigma)\ =\ \frac{1}{Z} \mathrm{exp}\ E(\sigma)\), where:
\[E(\sigma) = \sum_i \mathbf{h}_i (\sigma_i) + \sum_{i<j} \mathbf{J}_{ij} (\sigma_i, \sigma_j)\]
What’s significant about this change is that now we can score a given sequence on it’s overall likelihood.
EV energy scores correlate well with disease variation annotations and functional measurements

New applications also introduced by sequence-level modeling:
- deep mutational scanning experiments screen many genetic variants for an observed phenotype (ie does the protein glow more or less than other mutated proteins)
- more evidence that probabilistic models are generally useful

An update for the machine learning era: EVE



VAE models learn highly accurate ClinVar scores, even with only a couple hundred sequences per protein family.
Simple gaussian mixture on top of VAE probabilities gives ~90% correlation, ~99% AUC specific examples like TP53.
The state-of-the-art: masked language models


No sequence alignment needed: (pre-)training is over huge amounts of data–although model is relatively small (MM-15B param)
Brandes. et al (2023) paper demonstrates even without fine-tuning, ESM performs favorably compared with EVE
A bit unfair given the architectural context, but still clear differences using 650M parameter model:

Not shown–commensurate increase in prediction of mutational scanning effect datasets.
Embeddings from ESM can be easily fine tuned into highly accurate protein structural predictions



New applications of protein language models: forward de novo design


Note: people have been doing de novo design for ~20-30 years now, but it’s only in the last ~10 that it’s been possible with really extensive automation.
First paper: Ali Madani’s ProGen

Model is autoregressively trained from “control tags” (sort of like
First paper: Ali Madani’s ProGen

Most importantly, proteins designed using this method can actually be expressed in cells comparably to natural proteins – a big milestone
DL methods are enabling other applications in biology:

DNA Language models emerging (Benzegas et al. (2023), from Song lab at Berkeley): could be used to decode effect of regulatory DNA.
Builds on history of sequence modeling to predict transcription (Kelley lab at Calico, Enformer etc.)
Conclusions
Protein sequence modeling field has moved quite quickly in last five years.
Moving from simpler statistical models to more complex models, ultimately to learn energy functions / likelihood scorers.
Robust performance on variant scoring tasks–unclear what the field will do now that we have these strong general representation learners. So far analyses focused on protein-protein interaction, general prediction tasks (like stability).
